Portale "Ingegneria e Campanologia" - Autore - Sommario - Mappa del Sito - Home
![]() Strutturistica Chimica
Strutturistica Chimica
AREA I - ARTE TECNICO-SCIENTIFICA (ATS)
Cap. ATS-F02 - Chimica - Pag. ATS-F02.18
Gli argomenti trattati sono stati inseriti da Ing. Arch. Michele Cuzzoni nel 2012 - © Copyright 2007- 2025- e sono desunti dalla documentazione indicata in Bibliografia a fondo pagina
La determinazione strutturale tramite diffrazione di raggi X permette di ottenere un’immagine dettagliata della distribuzione elettronica all’interno di un cristallo.
Si assume che:
· Il cristallo ed anche una qualsiasi molecola o ione poliatomico siano costituiti da atomi ‘indipendenti’, ovvero che:
r(r) = r(xyz) = Sn r(r-rn)IAM = independent atom model
· Intorno al nucleo di ogni atomo, posizionato in
rn, la densità elettronica abbia distribuzione sferica r(r-rn);·
La trasformata di Fourier di r(r) sia: f(s) = f(sinq/l) = òr(r) exp(-ir.s)drr
(xyz) = 1/Vc ShSkSl F(hkl) exp[-2pi (hx+ky+lz)], ovvero: r(xyz) = 1/Vc òòòhkl F(hkl) exp[-2pi (hx+ky+lz)]dv*La Densità Elettronica è la Trasformata di Fourier dei Fattori di Struttura:
r(r) = T[F(r*)]Ovvero: L’intensità diffratta è l’Antitrasformata della Densità Elettronica: F(r*)= T-1[
r(r)] F(hkl) = |F(hkl)| exp(-ia)Se F(hkl) fosse sperimentalmente accessibile, basterebbe calcolare r(r) = T[F(r*)] per trovare la distribuzione di densità elettronica in cella.
Tuttavia:
Solo |F(hkl)| è noto, a non è sperimentalmente accessibile.La determinazione strutturale prevede la ricostruzione, per ogni hkl, della relativa fase, e, successivamente, il calcolo di
r(r) = T[F(r*)]Questo è il problema della fase, per cui esistono tecniche sofisticate non di soluzione, ma di approssimazione.
Siano dati:
F(hkl) = |F(hkl)| exp(i
a) F = |F|eia = A +iBr
(xyz) = 1/Vc ShSkSl F(hkl) exp[-2pi (hx+ky+lz)]Definiamo f = 2p(hx+ky+lz)
allora: r(xyz) = 1/Vc ShSkSl Fe-if
Fe-i
f = (A+iB)(cosf - isenf) = Acosf + Bsenf - i(Asenf - Bcosf)Dato che la somma va fatta su tutto lo spazio reciproco (tutti gli indici!), per ogni riflesso (hkl), c’è il suo equivalente di centrosimmetria (-h-k-l)
Tenuto conto che per i due equivalenti, A, B, cosf e senf hanno lo stesso valore assoluto, ma i termini in seno cambiano segno:
[sen(-x) = -senx; mentre cos(-x) = cosx]
e che:
A (somma di coseni) e cosf hanno lo stesso segno, e B (somma di seni) e senf hanno segni opposti
Nella somma su tutti gli hkl, le coppie centrosimmetriche fanno elidere il termine:
ShSkSl i(Asenf - Bcosf)Sommando pertanto su tutti i riflessi (escludendo i centrosimmetrici..):
r(xyz) = 1/Vc [F(000) + 2 Sh³0SkSl (Acosf + Bsenf)]ma, per A = |F|cos
a e B = |F|sena,r
(xyz) = 1/Vc [F(000) + 2 Sh³0SkSl |F|cos(f-a)]r
(xyz) = 1/Vc [ F(000) + 2 Sh³0SkSl |F|cos[2p(hx+ky+lz)-a] ]Se si conoscessero le posizioni approssimate e la natura di tutti gli atomi nella unità asimmetrica (ovvero, se si ha a disposizione un modello strutturale), si potrebbero calcolare |Fc| e
ac.Un confronto numerico tra fattori di struttura calcolati, |Fc|, ed osservati, |Fo|, può indicare se il modello riproduce, e con che precisione, i dati sperimentali, ovvero se il modello è ragionevolmente adeguato.
Analogamente, anche un confronto numerico tra le fasi calcolate,
ac, ed osservate, ao, può indicare se il modello riproduce, e con che precisione, i dati sperimentali, ovvero se il modello è ragionevolmente adeguato.
Quindi la precisione (non l’accuratezza!) di un modello strutturale viene valutato confrontando moduli di F secondo:
R =
S |(|Fo| - |Fc|)| oppure Rw = éS w[|Fo| - |Fc|]2 ù1/2S
|Fo| ë S w|Fo|2 ûCon le strumentazioni di oggi, tipici valori di R stanno tra 0.02 e 0.06 (2-6%). Valori più alti devono far sospettare errori o nel modello, o nella misura dei dati di intensità (o ambedue!)
Una distribuzione casuale di atomi in una struttura centrosimmetrica, una volta scalati gli |Fo| (statistica di Wilson) corresponde a R = 0.67 (67%).
Perché un modello ‘parziale’ (
posizione approssimata di solo alcuni atomi) possa eventualmente condurre al modello finale ‘corretto’, deve tipicamente possedere R tra 0.25 e 0.35. In queste condizioni, è possibile tentare di ‘espandere’ il modello, e trovare la posizione degli atomi mancanti.Una struttura cristallina e molecolare determinata per diffrazione è un’approssimazione della realtà, ovvero un modello che riproduce, in modo più o meno soddisfacente, i dati sperimentali.
Il modello strutturale non è mai provato, anche se, unito ad evidenze spettroscopiche, chimiche, etc., può essere considerato molto vicino alla realtà. Non è infrequente simulare modelli strutturali che sono più o meno sbagliati.
Quindi i passi logici per la formulazione di un modello strutturale completo sono:
· Formulazione di un modello, anche parziale, approssimato (Metodi Diretti, Funzione di Patterson, Scavengers, Monte Carlo, etc.)
· Espansione del modello a trovare gli atomi mancanti od eventuali errori nella sua formulazione.
· Miglioramento e finalizzazione del modello (Affinamento)
Espansione del modello tramite sintesi di Fourier
Siano dati i valori osservati di |Fo|
Sia disponibile un modello ‘parziale’ che pemette di calcolare |Fc| e
acrc(r) = ò |Fc| eiac e-if dV* densità elettronica calcolata (Fourier calcolata)
Contiene picchi laddove ho posizionato gli atomi nel modello
ro(r) = ò |Fo| eiao e-if dV* densità elettronica sperimentaleDovrebbe contenere picchi laddove stanno in realtà gli atomi
Se il modello di partenza è ‘abbastanza’ corretto,
ac » ao e:r
o(r) = ò |Fo| eiac e-if dV* (Fourier osservata)Contiene picchi laddove ho posizionato atomi nel modello ed anche in prossimità di atomi mancanti: ma è una mappa molto sporca..
r
o(r) - rc(r) = ò |Fo| eiao e-if dV* - ò |Fc| eiac e-if dV* = ò [|Fo| eiao - |Fc| eiac ] e-if dV* » ò [|Fo| - |Fc|] eiac e-if dV* (Fourier differenza)Contiene picchi solo in prossimità di atomi mancanti: è una mappa tipicamente molto più pulita della Fourier osservata. Fourier Osservata,
robs
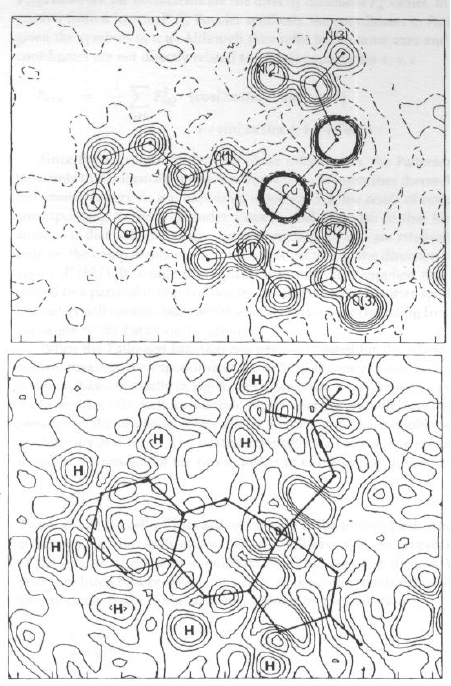
Fourier Differenza
Drobs-calcIn una mappa di Fourier Differenza, se il modello introdotto è approssimativamente corretto:
· Picchi positivi corrispondono a ‘atomi mancanti’ o ad atomi con Z troppo basso [C vs. S]
· Picchi negativi indicano che troppi elettroni sono stati messi in quel punto nel modello (atomo con Z troppo alto, P vs. N])
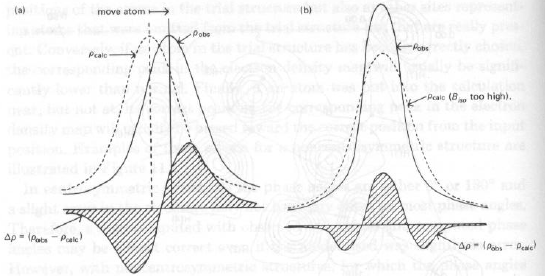
· Picchi positivi e negativi adiacenti suggeriscono di quanto va ‘spostato’ un atomo nel modello
Dx = -[¶Dr/¶x] / [¶2r/¶x2] = -(gradiente di Dr in x0) / (curvatura di r in x0)
dove x0 è la posizione di partenza,
oppure che il parametro termico assegnato è in errore.
Per Fobs Corretti e Fasi Corrette: Dr » 0
Per Fobs Corretti e Fasi Approssimate: Dr ¹ 0 informativa
Per Fobs Corretti e Fasi Incorrette: Dr ¹ 0 caotica poco informativa
Per Fobs Approssimati e Fasi Corrette: Dr ¹ 0 informativa
Per Fobs Approssimati e Fasi Incorrette: Dr ¹ 0 caotica poco informativa
Per Fobs Molto Approssimati e Fasi Corrette: Dr puo’ essere informativa
Contengono molte più informazioni le fasi che non le intensità, ma le fasi non si misurano e le intensità sì !
Tecnica numerica per trovare il miglior accordo tra un particolare modello sperimentale
e un set di dati sperimentali, quando siano presenti più osservabili che parametri da ottimizzare.Di per sé, non è un metodo propositivo, anche se alcuni dei suoi risultati possono suggerire modifiche sostanziali al modello.
In breve, si assume che i parametri migliori che descrivono il modello sono quelli che minimizzano la somma dei quadrati degli scarti tra quantità osservate e calcolate.
· Data una serie di n punti (di coordinate xi, yi), si può determinare qual è il polinomio a0 + a1x + … amxm (con n>m) che meglio descrive la funzione data per punti (problema lineare – esatto).
· Esiste la tecnica dei minimi quadrati pesati, che associa ad ogni osservabile un peso statistico (tipicamente inversamente proporzionale alla sua precisione).
· Tale metodo è facilmente adattabile a veloci calcolatori numerici, sia nella versione lineare che in quella non-lineare (approssimata).
In pratica, dato un gruppo di parametri P = {p1, ..pm}, i valori Fcalc(P) vengono confrontati con Fobs e minimizzata, al variare di P, la quantità:
Q =
Shkl w(hkl)[|Fo(hkl)|-|Fc(hkl)|]2dove w(hkl) = 1/
s2(|Fo|) (affinamento sugli F)oppure:
Q =
Shkl w(hkl)[|Fo 2(hkl)|-|Fc 2(hkl)|]2 (affinamento sugli F2)Quali sono i parametri in P?
Dato che |Fc| ha un’espressione analitica ben precisa, P contiene, per ogni atomo, x, y, z, B (modello isotropo, 4 parametri)
o
x, y, z, b11, b22, b33, b23, b13, b12 (anisotropo, 9 parametri)Occasionalmente, per alcuni atomi, in presenza di occupanze frazionarie, si può affinare un parametro di occupazione o disordine..
Inoltre, c’è sempre un fattore di scala (dal Wilson plot, ma affinabile..) tale che |Fc| sia approssimato da k|Fo|
Quindi, in generale, i parametri affinabili sono (9N+1)
Per una struttura semplice come: [Co(NH3)5(NO2)]Br2 Ci sono 26 atomi (di cui 15 idrogeni, tipicamente isotropi..)
Numero di parametri: 11 x 9 + 15 x 4 + 1 = 160
Per avere sufficiente accuratezza, servono tipicamente dai 7 ai 12 dati sperimentali per parametro
Ovvero: circa 1600 riflessi osservati (con |Fo| ¹ 0)
Matematicamente: bisogna trovare il minimo (assoluto, non relativo!) di una funzione Q di 160 variabili reali!
Ovvero, bisogna mettere a zero la derivata di Q per ogni parametro
pi (garantendo la curvatura positiva…)Questo processo non è per niente lineare, dato che entrano in gioco funzioni trigonometriche, esponenziali, etc. e quindi non può essere risolto esattamente, senza un modello di partenza.
E’ comunque possibile linearizzarlo se sono noti i valori approssimati
po e si vogliono trovare i valori Dp dell’espansione di Taylor al prim’ordine. In questo caso la soluzione approssimata sarà ottenibile attraverso diversi cicli di minimi quadrati, fino a convergenza, ovvero fino a quando i Dp sono dell’ordine della loro incertezza statistica (esd)In :
D|Fc| = ¶|Fc|/¶x1 Dx1 + ¶|Fc|/¶y1 Dy1 + …+ ¶|Fc|/¶b33,n Db33,nSono i
Dpi che vengono calcolati ciclo per ciclo come incognite..
Per favorire il processo di convergenza dei minimi quadrati o per imporre particolari condizioni geometriche, è possibile condizionare il processo di affinamento limitando il numero o il campo di variabilità dei parametri
Constraint: relazione matematica esatta che fissa un parametro o mette in relazione parametri diversi.
Esempi:
· Per un atomo in su un asse binario lungo y, x e z sono fisse, p.es. a 0, 0.25 o 0.50 (a seconda del gruppo spaziale e dell’origine scelta)
· Per un atomo su un piano diagonale xxz, x e y devono essere uguali
· Per sei atomi di un fenile, si può imporre che la sua simmetria sia
D6h, e, dei 6x3(xyz) = 18 parametri, solo 7 siano indipendenti (xyz e Rx,Ry,Rz del centro di massa + dC-C): modello a corpo rigido· Per atomi vicarianti (FeII e MgII in MSiO4), occ(Fe) = [1.0 - occ(Mg)]
Restraint: Osservazione sperimentale aggiuntiva che deriva dall’analisi di banche dati, modelli esterni, spettroscopia (NOE, I.r.) che limita il campo di esistenza per alcuni parametri (pi ± Dpi) o di una loro funzione d(pi,n)±Dd(pi,n). Matematicamente, aumenta il numero delle equazioni (una per ogni osservazione), ma necessita di essere ulteriormente ‘pesata’.
In questo caso:
QTOT = Shkl w(hkl)[|Fo(hkl)|-|Fc(hkl)|]2 + WSp wp[do-dc]2
QTOT, funzione costo complessiva, può contenere termini geometrici, energetici, spettroscopici, etc.
Esempi:
· Distanze tra atomi; angoli di legame.
· Parametri termici isotropi (variazione all’interno di una certa s)
Durante il processo di costruzione del modello e di affinamento:
· Il fattore di accordo Rw deve scendere (minimizzazione di Q).
· Gli atomi devono muoversi di ‘poco’.
· Il fattore Biso deve, di norma, essere omogeneo con la connettività.
· I fattori
bij non devono essere ‘particolarmente’ anisotropi (o dare ellissoidi immaginari!).· Passo dopo passo, la mappa di Fourier Differenza deve essere sufficientemente piatta.
· Si devono evitare falsi minimi (minimi relativi!)
Alla fine del processo computazionale:
Non ci sono test universali ma:
1. I valori |Fo(hkl)|-k|Fc(hkl)| devono scostarsi da |Fo(hkl)| di circa
s|Fo(hkl)| o poco più.2. La mappa di Fourier Differenza Finale deve mostrare picchi e valli in Dr comparabili con s(Dr).
3. Qualsiasi anomalia geometrica nella molecola o nelle interazioni di impaccamento deve essere valutata, commentata e giustificata.
Le esd di ogni parametro hanno senso statistico solo per errori casuali, in assenza di errori sistematici, molto comuni. Spesso rappresentano solamente la precisione, non l’accuratezza. Misure interlaboratorio (sponsorizzate IUCr, ICDD, o altro) sullo stesso campione hanno evidenziato come l’accuratezza possa essere 3 o più volte inferiore! Se la distribuzione degli errori casuali è normale, allora la teoria dei minimi quadrati stima che:
· Due valori che differiscono di meno di 2 esd, sono uguali al 95% di probabilità;
· Due valori che differiscono di meno di 2.7 esd, sono uguali al 99% di probabilità;
Comunemente, si confrontano 2 valori con la regola: Se differiscono di meno di 3 esd, non sono statisticamente differenti. 1.560(7) non è significativamente diverso da 1.542(3)
Corollario: per distinguere due valori vicini, devo essere in possesso di determinazioni strutturali di grande accuratezza (tipicamente 0.001 Å per M-M;< 0.01 Å per C-C, C-N, e <1° per gli angoli di legame). 1.560(3) è significativamente diverso da 1.542(2)
· Determinazioni strutturali grossolane suggeriscono la stechiometria, la connettività, la forma molecolare ma solo raramente parametri geometrici accurati.
· Tipicamente, determinazioni strutturali grossolane si possono ottenere da mancata cura nella preparazione del campione, o nella strategia di raccolta ed analisi dati.
Errori sistematici comuni:
· Le curve dei fattori di scattering si assumono generalmente sferiche, senza dipendenza vettoriale. Recenti studi teorici e sperimentali hanno dimostrato che atomi legati non sono sferici ma la loro deformazione di densità elettronica verso i legami non è trascurabile. (Promolecola S(IAM)==> Molecola Reale).
· Il moto degli atomi in un cristallo può essere correlato (p.es. a vibrazioni collettive di reticolo) o anarmonico (non bastano i bij).
· Disallineamento strumentale, assorbimento, contaminazione della radiazione…
· Effetto Renninger (diffrazione multipla): può capitare che un raggio riflesso molto intenso sia in grado di dare ulteriore diffrazione. Il raggio uscente sporca la raccolta dati in termini di intensità.
Portale "Ingegneria e
Campanologia" -
Autore -
Sommario
- Mappa del Sito -
Home
Bib-TS-081 - Prof. N. Masciocchi - Dispense del Corso di Laurea in Chimica - Insegnamento di strutturistica chimica
Bib-TS-082 - C.Hammond - The Basics of Crystallography and Diffraction - Ed. International Union of Crystallography and Oxford University Press, 240 pg. (Ed. italiana: Zanichelli)
Bib-TS-083 - J.P.Glusker & K.N.Trueblood - Crystal Structure Analysis: A Primer - Oxford University Press, 220 pg. (non tradotto).