Portale "Ingegneria e Campanologia" - Autore - Sommario - Mappa del Sito - Home
![]() Analisi chimiche per materiali e metalli
Analisi chimiche per materiali e metalli
AREA I - ARTE TECNICO-SCIENTIFICA (ATS)
Cap. ATS-F03 - Chimica - Pag. ATS-F03.09
Gli argomenti trattati sono stati inseriti da Ing. Arch. Michele Cuzzoni nel 2012 - © Copyright 2007- 2024 - e sono desunti dalla documentazione indicata in Bibliografia a fondo pagina
-
Caratterizzazione cromatografica dei materiali glicerolipidici e delle cere
-
Caratterizzazione cromatografica dei materiali polisaccaridi
Eccellente tecnica sia qualitativa che quantitativa.
Le tecniche cromatografiche sono tecniche di SEPARAZIONE: infatti permettono di separare e successivamente determinare gli analiti presenti in miscele anche complesse.
PRINCIPIO DI SEPARAZIONE:
gli analiti si distribuiscono in due fasi, ovvero due parti assolutamente immiscibili tra loro e dunque separate, di solito per il loro diverso stato (solido, liquido, gassoso).E’ però efficiente solo per alcune classi di composti, perché altre specie si confondono perché si mischiano ad altre. Infatti gli analiti vengono a contatto con le 2 fasi e si possono ripartire (se le due Fasi sono liquide) o si adsorbono (quando una delle due fasi è solida) in maniera differenziata nelle 2 fasi a seconda della loro affinità (cioè solubilità) con esse. Infatti per separare efficacemente bisogna scegliere bene le 2 fasi, in modo da ottenerne una mobile che si staccherà più facilmente dall’altra, che invece rimarrà ferma. Per questo si distinguono una fase mobile (FM) e una fase stazionaria (FS).

FASE STAZIONARIA (FS)
= o è un solido o è un sottile film liquido stabile che ricopre la superficie interna del contenitore (di silice); si tiene unita e sigillata .FASE MOBILE (FM) = Si trova all’interno del contenitore ed è liquido o gassoso e all’interno del contenitore riesce comunque a fare interscambi con l’esterno perché la FS non è completamente sigillante.
Nella tecnica, il contenitore è come una colonna, cavo sia sopra che sotto. La FM attraversa questo tubo (rivestito internamente di FS) attraverso un sistema di pompe.
Di solito il passaggio è più facile per un liquido che per un gas perché le molecole rimangono più compatte e vicine tra loro, quindi il passaggio tramite pompe è facilitato.
Nella colonna di FS mettiamo dentro l’analita con la FM; se predispongo bene la composizioni delle due fasi, mi aspetterò che l’analita dimostri più o meno affinità ionica con esse (cioè se ha simile struttura: polare, apolare, parzialmente polare). Valutando i due casi:
- Se l’analita è simile alla FM → l’analita si lega alla FM ed esce velocemente
- Se l’analita è simile alla FS → l’analita forma legami temporanei con la FS quindi rallenta la sua discesa
Soprattutto il secondo punto è importantissimo perché l’unico discrimine per distinguere gli analiti è il loro tempo di ritenzione all’interno della colonna.
Di conseguenza, è una tecnica che dipende direttamente dalla struttura chimica dei vari agenti coinvolti, e bisogna anche considerare, all’interno dei macro gruppi di affinità, anche forze attrattive di minore entità (cioè sono tutti affini, ma comunque a diversi gradi); infatti composti molto simili potrebbero uscire dalla colonna ancora insieme. Per ovviare a ciò si può riscaldare il tutto, perché la temperatura fa differenziare nel comportamento dei composti anche molto simili.
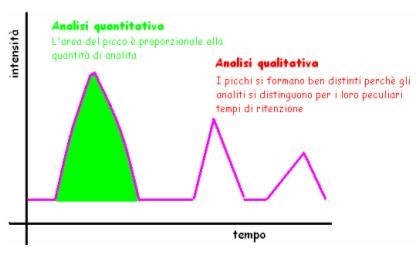
Alla fine del loro percorso nella colonna, tutto ciò che ne esce (FM e analita) viene registrato da un rilevatore, che registra in modo preciso i tempi dei vari gruppi di composti, trasformando questi dati in un CROMATOGRAMMA, che registra le uscite di materiale come picchi, più o meno ampi a seconda della loro quantità. In teoria ogni picco dovrebbe corrispondere ad un analita, perché dovrebbero rimanere tutti perfettamente isolati, se tutto il lavoro è stato svolto con cura.
L’informazione QUANTITATIVA viene ricavata dall’area o dall’altezza del picco, perché sono proporzionali alla concentrazione dell’analita:
A = k . C (non è la legge di Lambert Beer perché riguarda l’area!!), vale anche per l’altezza.
Per preparare le rette di taratura, per ogni analita dispongo 4 soluzioni standard e con i risultati ottenuti costruisco una retta. C’è un programma che le costruisce automaticamente, ma questo procedimento va fatto elemento per elemento. Con più analiti posso fare le analisi per tutti questi elementi tutte insieme, basta solo conoscere le concentrazioni.
Precisazione: La linea del cromatogramma non segna mai l’intensità 0 perché comunque gli analiti scendono separati, ma col flusso continuo di FM che fluisce continuamente.
Permettono di separare miscele complesse di composti di separazione: avviene sfruttando un diverso comportamento chimico-fisico che ciascun componente ha rispetto a un sistema di fase fissa (fase stazionaria), costituita da particelle solide generalmente contenute in una colonna, e di fase in movimento (fase mobile), costituita da liquido o gas che fluisce nella colonna.
Interazioni tra componenti da separare e le due fasi: responsabili del diverso tempo necessario a ciascuno di essi per emergere da colonna.
Composti separati: rilevati da specifici rivelatori (spettrometro di massa).
A seconda di tipo fase mobile usata, si hanno diverse cromatografie: liquida se fase è mobile e liquida, tecniche gas cromatografiche se fase è gassosa.
Metodica senza pretrattamento campione: con pirolisi interfacciata alla GS-MS si ottengono programmi altamente specifici di tutte le sostanze organiche presenti; i migliori risultati si hanno quando all’atto della pirolisi si fa seguire la reazione di derivatizzazione in situ e si aggiunge opportuno agente derivatizzante direttamente nella cella di pirolisi.
Vengono resi volatili gli analiti con funzionalita acide: liberati durante pirolisi e analizzabili con GC-MS permette di identificare polimeri di sintesi.
E' possibile distinguere tra uovo e olio siccativo basandosi su quantità relative tra acidi carbossilici di impossibile distinzione tra vari tipi di oli siccativi: conservazione di trigliceridi in acidi grassi liberi non è completa per effetto di reazione secondarie.
Approccio più usato per caratterizzazione materiali proteici: idrolisi acida proteine seguita da determinazione profilo amminoacido tramite diverse tecniche cromatografiche.
Il passaggio piu critico è l'analisi dell'idrolisi: è opportuno trovare un giusto compromesso tra l'efficacia di idrolisi dei legami peptidici più stabili e necessità di ridurre al minimo le perdite di amminoacidi chimicamente meno stabili.
Alcuni cationi inorganici e pigmenti interferiscono nella determinazione di amminoacidi che danno luogo a formazione di complessi stabili; con alcuni di essi si ha interferenza minimizzata attraverso la fase di purificazione dell'acido idrolizzato: essa è basata sull'utilizzo di resine a scambio cationico.
Tecniche utilizzate:
* CROMATOGRAFIA SU STRATO SOTTILE (TLC):
amminoacidi rivelati attraverso spettrofotometria di fluorescenza molecolare.* CROMATOGRAFIA LIQUIDA AD ALTA PRESSIONE (HPLC):
amminoacidi separati in fase inversa e rivelati per spettrofotometria UV-VIS o di fluorescenza molecolare.* CROMATOGRAFIA A SCAMBIO IONICO (IC):
amminoacidi separati tramite cromatografia a scambio ionico.* CROMATOGRAFIA IN FASE GASSOSA (GC):
gas cromatografia accoppiata a spettrometria di massa (GC-MS) e la gas cromatografia con rivelatore a ionizzazione di fiamma (FID) sono tecniche piu usate; è necessario volatilizzare amminoacidi per renderli volatili.Dal profilo dell'amminoacido è possibile identificare il tipo di proteina.
Principio: FM gas (inerte, come elio e azoto) e FS solida o liquida. Il campione deve essere un gas o un liquido volatile. Se non lo è,il campione deve essere trasformato in un suo derivato liquido, noto e volatile (processo di derivatizzazione). Viene reso volatile con una sostanza disciolta insieme: si forma così una nuova specie chimica, in cui però posso distinguere gli analiti.
In queste condizioni le uniche sostanze indagabili sono quelle organiche (leganti pittorici, ma anche resine, cere, gomme vegetali…).
Parametri che ottimizzano la separazione:
- Tipo di FS.
- Programma di temperatura a cui è sottoposta la FS.
Svantaggi:
- Pre-trattamento del campione.
- Numero elevato di campioni standard.
Ci sono due modifiche di questa tecnica che permettono di aumentare la sensibilità della tecnica:
Con questa analisi si può partire da sostanza anche solide, anche se non gassificabili o solubili (come polimeri reticolati come olii e resine); non faccio la derivatizzazione, ma la pirolisi = scaldare fino a 1000°C; così si ottiene un gas che viene immesso nella colonna. Questo metodo però va bene solo per le miscele più semplici, perché quelle complesse non sempre si sciolgono bene.
Purtroppo bisogna considerare che, oltre all’analisi di soli composti semplici, ha anche un costo elevato.
Principio: si separano gli analiti grazie all’impatto con un fascio di elettroni, che frammentano il campione, ma non in maniera casuale: infatti si ripartiscono tra gruppi di molecole abbastanza caratteristici (per la loro carica). E’, come dire, una frammentazione caratteristica.
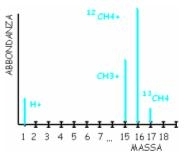
Esempio di analisi del metano: CH4 viene frammentato in CH4+ + CH3+ + H+ . Poi queste molecole caratteristiche vengono rintracciate singolarmente nel cromatogramma. Così possiamo confermare le nostre ipotesi sulla composizione del materiale in questione.
In più è meglio spendere qualche parola su questo particolare cromatogramma, che viene sottoposto all’ANALISI CHEMIOMETRICA. Le sostanze organiche sono più o meno proteiche o lipidiche (posso rivelarlo con reazioni con composti ad hoc): in genere per ogni campione ho in genere tutti e due i tipi di sostanza organica e li rilevo inserendoli in un diagramma, che tiene conto anche dell’invecchiamento. Con delle elaborazioni statistiche dell’analisi chemio metrica, io posso rappresentare un campione con un punto; con molti campioni ottengo una massa di puntini da cui posso trarre un’affidabile via di mezzo.
Da una parte però le cose si complicano perché si abbassa il limite di rivelabilità: nei leganti abbiamo concentrazioni bassissime di sostanza organica. In più c’è anche un altro problema sempre collegato all’analisi di sostanze organiche, cioè il loro invecchiamento; e quindi tenere conto anche delle rette di taratura adatte. Il problema è che infine, tutti questi accorgimenti alterano la validità dei risultati.
Principio: ripartizione degli analiti, basata di solito sul principio dello scambio IONICO (“cromatografia ionica”), basata su una FM liquida (o solvente o miscela di solventi) e una fase stazionaria solida nella colonna.
Per trasportare il campione attraverso le colonne servono PRESSIONI ALTE: questo perché ci vuole forza per far passare la FM liquida attraverso la FS solida. Si parla quindi di “cromatografie liquide ad alta pressione” (HPLC=high pressure liquid chromathography)
Campione: pochi mg ovviamente solubilizzati; quelli utili all’analisi alla fine sono poche decine di μl.
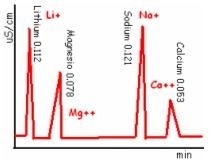
Applicazioni: analisi di Sali solubili (come efflorescenze) divisi in cationi e in anioni, spesso rintracciabili nei materiali lapidei.
L’analisi della presenza e della quantità di cationi e anioni è fondamentale per valutare lo stato di degrado dei materiali lapidei. Infatti i Sali solubili sono fenomeni di degrado più frequenti delle malte e delle murature; possiamo anche analizzare efflorescenze e cripto efflorescenze.
Principali:
- Cationi +:
calcio, magnesio, sodio, potassio, ammonio- Anioni - :
nitrati e nitriti, solfati, cloruri, fosfatiIl campione solubilizzato NaCl viene ionizzato in Na+ e Cl-. i campioni vengono macinati e aggiunti ad acqua de-ionizzata (acqua passata attraverso resine che trattengono ioni). Il grafico che poi risulterà al passaggio avrà in ascissa il tempo e in ordinata l’intensità, misurata in conducibilità.
Parametri che ottimizzano la separazione sono ovviamente il tipo di FS e FM scelti.
Vantaggi: Si presta bene all’automazione, perché il campione può venire iniettato con una siringa nel cilindro, evitando altre manipolazioni che potrebbero alterare in risultati.
Svantaggi: non è una tecnica di speciazione (troviamo sì solfati, ma sono di Ca o di Mg?), anche se posso cercare di combinare a logica anioni e cationi.
E' fondata su idrolisi (in genere basica condotta in KOH alcolico) materiale glicerolipidico e cere, su successiva derivatizzazione e determinazione degli acidi grassi, alcol e idrocarburi e con analisi mediante GC-MS; le reazioni di derivatizzazione portano alla
FORMAZIONE DI ETERI ED ESTERI VOLATILI:* Metilici
* Isopropilici
* Trimetilsilanici
Differenziazione di vari materiali glicerolipidici attraverso la valutazione di alcune grandezze caratteristiche:
- rapporto tra percentuali relative di acido palmitico e acido searico (permette distinguere tra olio di lino, olio di noci e olio di papavero);
- rapporto tra percentuali relative di acido azelaico e acido palmitico insieme a somma percentuali di acidi carbossilici e permette di distinguere olio siccativo da tempera ad uovo.
Gas cromatografia: accoppiata a spettrometria di massa è la tecnica piu diffusa per caratterizzazione i materiali resinosi.
Generalmente prevede lo stadio preliminare di idrolisi, matrice ridotta nelle sue componenti glicosidiche e la successiva determinazione dei monosaccaridi e degli acidi uronici liberi tramite procedura cromatografica.
Idrolisi: si effettua in condizioni acide controllate; il problema principale risiede nelle rese quantitative e qualitative dei componenti della matrice polisaccaridica originale; a seconda delle condizioni sperimentali usate si può avere degradazione o interconversione delle componenti glicosidiche.
Determinazione del gas con cromatografia zuccheri: è necessario uno stadio preliminare di derivatizzazione dei monosaccaridi e degli acidi uronici per renderli meno polari e aumentarne la volatilità.
Cromatografia liquida ad alta pressione è molto usata per studio dei carboidrati.
Portale "Ingegneria e
Campanologia" -
Autore -
Sommario
- Mappa del Sito -
Home
Dispense tratte da: http://doc.studenti.it/download/skip/chimica-ambiente-beni-culturali_1.html (nel 2012, ora il sito non è più esistente)
Bib-TS-086 - M. Matteini, M. Moles, la Chimica nel restauro, Nardini editore, Firenze
7 - M. Matteini, M. Moles, Scienza e restauro, Nardini editore, FirenzeBib-TS-088 - L. Appolonia, S. Volpin, Le analisi di laboratorio applicate ai beni artistici policromi, casa editrice il Prato, Padova